
集思慧远又一QTL软件分析来啦!今天小编要跟大家分享的是WinQTLCart。
WinQTLCart是北卡罗来纳州立大学发布的一款在windows下运行的QTL软件,目前最新版本为V2.5_011。WinQTLCart可以适用BC1,DH,RIL,F2等群体,但是不能使用CP群体进行分析,可以使用IM(区间作图法),CIM(复合区间作图法),MIM(多区间作图法)进行QTL的分析,这个软件运行时间比较长,对于标记数量过多的分析,最好不要在运算的同时,运行其它大型程序,可能会引起死机。



首先下载软件WinQTLCart,下载网址:https://brcwebportal.cos.ncsu.edu/qtlcart/WQTLCart.htm。
鼠标双击WinQTLCart.exe文件即可运行。


1、打开软件,查看分析页面情况。

New:从原始文本文件创建一个新的源数据文件。
Simu:打开模拟数据对话框。
Dsum:汇总活动源数据文件的统计信息并显示在“文本”窗口中。
Result:在“图形”窗口中查看结果文件,请参见图形窗口。
DIR:设置默认工作目录。
DrawChr:显示并打印当前活动的.MCD文件中染色体的图形显示。
IM:interval Mapping,区间作图法。
CIM:复合区间作图法。
MIM:多区间作图法。
NotePad:打开记事本,使用记事本作为方便的文本编辑器来帮助格式化源数据文件。
2、染色体图显示。选择Tools->DrawChrom可以展示遗传图谱信息。然后调整图片进行,最后输出。
3、点击Source Files,然后打开输入数据文件*.mcd文件。
4、Importing files功能,除非您正在处理的文件已另存为WinQTLCart映射源数据文件(具有.MCD扩展名的文件)或已经为.MCD格式,否则您需要将它们导入WinQTLCart。WinQTLCart将读取文件,验证数据格式,并自动将文件保存为.MCD格式。
5、WinQTLCart可以从以下应用程序导入文件:MapMarker/QTL .MAP(图谱文件),.MAS(图谱文件),.RAW输入数据,或者QTL Cartographer的.INP图谱和数据结合文件,或者.MAP图谱文件,.CRO输入数据;或者excel文件


 注意:如果WinQTLCart显示一条错误消息,指出文件格式无效或无法识别,则扩展名可能是错误的,或者文件的格式使其无法在WinQTLCart中使用。在记事本中打开文件,并将其与WinQTLCart目录中的示例文件之一进行比较。
注意:如果WinQTLCart显示一条错误消息,指出文件格式无效或无法识别,则扩展名可能是错误的,或者文件的格式使其无法在WinQTLCart中使用。在记事本中打开文件,并将其与WinQTLCart目录中的示例文件之一进行比较。
6、导入源数据文件,选择源文件类型,选择inp类型文件作为输入文件,然后点击下一步。

7、点击下一步,分别导入map图谱文件和cross数据文件->给源文件命名->设置目录->然后点击完成,导入文件完成。关于输入WinQTLCart将创建的源数据文件的文件名,也无需指定扩展,WinQTLCart会解决这个问题,如果选中“从交叉数据文件推断图谱信息”,WinQTLCart将使用Emap函数从交叉数据文件推断图谱信息。

8、如果使用Emap功能推断图谱信息,则将弹出Emap参数设置对话框。
单击按钮更改Emap功能的随机种子。链接映射方法的值可以为10、11、12或13。隔离测试大小可以为0.01到0.20。链接测试大小可以为0.15到0.49。现在,置换功能不起作用。作图功能可以是Haldane或Kosambi。目标功能可以是SAL或SAR。

9、使用“锚标记”设置控制组设置锚标记。单击以选择锚标记号。单击按钮参数以打开锚定标记参数集的窗口,选择每个标记的标记标签,染色体和在染色体上的位置(当前锚标记)。单击确定按钮以完成参数设置。

如果导入成功,您将看到一个对话框,提示文件已成功导入并且源数据文件已保存。如果导入无效,则可能是文件格式不符合WinQTLCart期望的标准格式的原因。对于INP格式,如果使用Emap推断图谱信息,则需要一个交叉数据文件。
10、WinQTLCart的源数据文件具有.MCD扩展名。.MCD文件是遵循特定格式的文本文件,其中包括WinQTLCart用于QTL分析的所有信息。尽管可以在WinQTLCart中打开其他文本和结果文件,但是“窗体”窗格中仅显示.MCD文件信息。即使打开或切换到文本或结果文件,WinQTLCart仍将继续显示在“表单”窗格中查看的最后一个.MCD文件。
11、对数据的总结页面。

最左边的部分是对数据的总结介绍,中间的是对数据的查看和修改参数设置,最后是对数据进行分析的参数信息。选择Interval Mapping(区间作图法),然后点击GO,进行QTL分析界面。
12、在工作窗口可以查看导入的数据信息。

--type:遗传图谱展示方式,position指的是展示遗传图谱上每个标记的遗传距离排序;interval表示数字用于标记后的间隔距离。这意味着最后一个数字(仅最后一个数字)应为零
--function:1代表Haldane;2代表Kosambi;3代表Fixed;
--units:遗传距离的单位(cM);
cM(centiMorgan):很小,100厘摩= 1M。
M(Morgan):每个减数分裂平均发生一次交叉的距离。
重组率(Recombination frequency):两个标记之间发生的交叉事件的百分比。;
--chromosome:源数据中总连锁群的数量;
--maximum:13;指示所有源数据的染色体最大标记数。
--named:Yes,表示标记具有名称;No,绝不意味着标记将没有名称。;
--start and --end:使用这些标记可以开始和结束所有染色体的标记位置数据;
--Chromosome:指示染色体名称,该染色体的标记位置数据将从下一行开始;
#bycross:表示交叉信息即将开始;

--SampleSize:表示样品数量
--Cross:指示交叉类型配合设计的代码;
--traits:指示源数据的特征编号;
--otraits:指示源数据的其他特征编号。其他特征(也称为分类特征)是具有定性或分类值的特征,例如性别;颜色等等。其他特征可以用作因素可以在回归分析中“回归”。这意味着将对感兴趣的定量性状进行分类性状回归,并将残差用作分析中的表型。
--missingtrait:表示缺少特征值的符号;
--case是或否。是,表示所有比较均取决于大小写。绝不表示将个人,标记和特征的所有名称都转换为小写以进行比较。
–TranslationTable:带有表(数据的后6行)将定义标记基因型数据的翻译方式。
13、以下关于IM分析的参数设置,默认设置将提供最佳的全方位参数设置,尤其是对于初学者来说。
单击结果文件,以选择位置并命名分析完成后将创建的.QRT文件。
单击OTraits以输入其他特征编号或编号范围。WinQTLCart将在分析中将它们用作辅助因素。(WinQTLCart的默认值为“无OTraits”)。
Walk speed(cM):1cM,提高数值意味着精度较低,但分析所需的时间更少。降低数值会产生更精确的结果,但会花费更多时间。
整个数据集设置同样的步行速度值。单一步行速度建立了一致的规范,可以根据该规范绘制数据。如果更改两次贴图运行之间的步行速度,则图形显示将倾斜。
Chromosome Selection:选择一个或所有染色体进行分析。
Trait Selection:选择一个或所有性状包括在分析中。
Threshold:LOD阈值设为3。
Permutation Times:最好选择1000及以上,比较可信,但是运行时间较长。
设置完成后,点击OK/START进行分析。

14、经过分析,会产生以下文件:
1)创建一个QTL映射结果文件(* .QRT),并在“图形”窗口中将其打开
2)使用EQTL函数创建QTL摘要信息文件。
3)在主窗口的“数据”窗格中显示一个确认框,询问您是否要显示QTL摘要信息。
要查看“图形”窗口,请通过单击主窗口工具栏上的“结果”按钮来打开WinQTLCart结果文件(扩展名为.QRT)。将打开“图形”窗口,其中显示了结果文件数据的图形概述。
找出QTL的位置。超出阈值线的图峰是QTL的位置,可以一次加载多个结果文件并进行比较。底部的较小图形是QTL效果窗口,显示加性或显性效果或R2或TR2或S值。阈值线以上的峰表示QTL。图上的高LOD值表示良好的QTL候选对象。
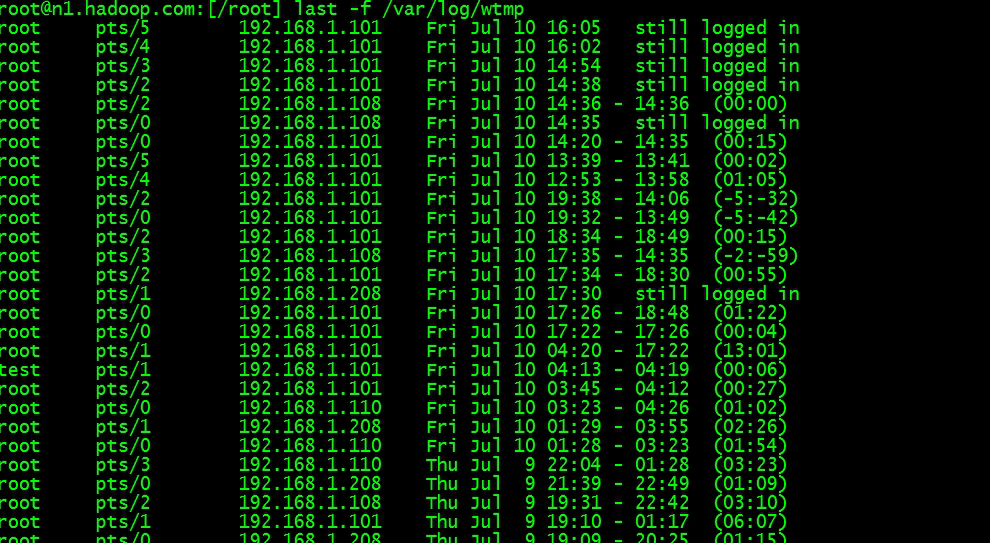
右键点击窗口,设置参数。
选择设置>图形中的跟踪坐标或单击。在屏幕上移动光标时,请注意,图形坐标显示在图形的右上角。
QTL编号:设置要在文件中显示的QTL数量。
遗传力:需要一个介于0.0到1.0之间的值。默认值为0.8。
Vd / Va:显性方差/加性方差
Vi / Va:上位方差/加性方差
分析的结果在窗口展示:
ADDitive:加性效应;
Dominance:显性效应;
R2:贡献率;

今天的分享到这就结束了,还想学习哪方面知识与技能?科研还有哪些困惑疑问?记得留言告诉我们哦~

扫码关注
有趣的灵魂在等你

▼往期精彩回顾▼
QTL Icimapping系列又出新!看看有啥新教程
大规模线粒体测序,揭示小龙虾的全球入侵之路
集思慧远又双叒叕出新的分析教程啦!









 京公网安备 11010802041100号
京公网安备 11010802041100号