前言
Liftover是UCSC中用于基因组版本之间转换的一个工具,既可以做某一物种内基因组版本的转换,还可以做物种间基因组版本的转换。
UCSC提供了两种操作方式,既提供了网页版,还提供了linux上的版本。
1、网页上操作相对来说比较简单,首先需要选择几个重要的参数。

包括输入文件的物种,基因组版本号,输出文件的物种,版本号信息,如果下拉菜单中没有此物种的信息,则证明不能转换。

这个参数为映射的最小比率,默认为0.95。
输入文件要求是bed格式或者chrN:start-end格式,既可以复制文件内容到界面的输入框中,也可以提交bed格式的文件或chrN:start-end格式的txt文件。

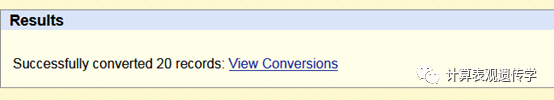
转换成功之后会提示类似以下内容的信息,点击蓝色链接可以下载转换后的文件。

2、如果需要转换的文件较多,可以下载linux版本的liftover软件进行批量处理。使用以下命令可以下载liftover软件:
wgethttp://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/liftOver
如果你需要在linux系统操作liftover 这个工具,同时还需要下载坐标注释文件http://hgdownload.cse.ucsc.edu/downloads.html#liftover。我下载的是hg38版本转hg19版本http://hgdownload.cse.ucsc.edu/goldenPath/hg19/liftOver/hg19ToHg38.over.chain.gz。

-minMatch=0.N map的最小比率,默认为0.95。
Hg38_CpG.bed是转换成功的文件,而unmap是转换失败的信息。
3、liftover现在也可以在R版本上进行基因组版本的转换了,操作起来也十分简单。以下命令可以加载liftover的R包。
source("https://bioconductor.org/biocLite.R")
biocLite("rtracklayer")
library(rtracklayer)
使用Granges参数把数据转化为固定的格式
Hg19
Hg19
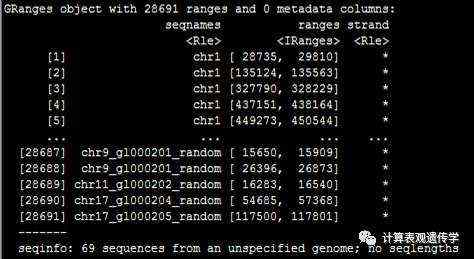
使用下面的命令可以进行基因组版本的转换。
hg38 = liftOver(hg19, chain)
hg38 =unlist(hg38)
之后把结果读出来就可以了。
希望这次报道对大家有所帮助!





 京公网安备 11010802041100号
京公网安备 11010802041100号