CSV/TSV作为数据科学和组学分析的基本格式,其实本质上均为txt格式的表格,CSV是按逗号分隔,TSV是以制表符分隔的表格。这两种格式数据格式应用非常广泛。比较常用的处理软件包括:
Excel为代表的电子表格软件
Notepad++/Edit-plus等文本编辑器
sed/awk/cut等Shell命令
各种编程语言,例如 R、python等。
备注:sed/awk/cut等Shell命令不适合含有标题行的CSV格式,当然这些命令的操作非常快。
csvtk软件用GO语言编写,是一个支持多平台(Winodws/Mac/Linux)的小工具,支持格式除了这两种格式外还可以处理gzip压缩的格式。无需解压即用。27个子命令支持管道组合使用,支持简单的出图。
本文主要参考软件官方教程 https://bioinf.shenwei.me/csvtk/usage,并将方法应用于扩增子分析中常用的特征表(otutab.txt)、元数据(metadata.tsv)和物种注释(taxonomy.txt)。
软件主页:https://github.com/shenwei356/csvtk
软件下载:https://github.com/shenwei356/csvtk/releases/
各系统版本都下,下载解压即可使用。
可选使用conda安装
conda install csvtk
# 信息
headers 打印标题行,如果表格列比较多,首先查看列名方便后续操作
stats 基本统计分析
stats2 对指定的列进行基本统计,注意需要是数值列
# 格式转化
pretty 转为美观、可读性强的格式,用于打印
csv2tab 转CSV为TSV
tab2csv 转TSV为CSV
space2tab 转空格分割格式为TSV
transpose 转置CSV/TSV 这一个步骤往往看看出来工具好不好用
csv2md 转CSV/TSV为makrdown格式,方便我们多平台排版,发文# 集合操作
head 屏幕打印表格前面的内容,可以指定行数
sample 按比例随机采样 对行进行抽样
cut 选择特定列,支持按列或列名进行基本选择、范围选择、模糊选择、负向选择(最常用命令之一,非常强大)
uniq 无须排序,返回按指定(多)列作为key的唯一记录
freq 按指定(多)列进行计数(常用)
inter 多个文件的交集
grep 指定(多)列为Key进行搜索(最常用命令之一,可按指定列搜索)
filter 按指定(多)列的数值进行过滤
filter2 用类似awk的数值/表达式,按指定(多)列的数值进行过滤
join 合并多个文件(常用)# 编辑
rename 直接重命名指定(多)列名
rename2 以正则表达式重命名指定(多)列名
replace 以正则表达式对指定(多)列进行替换编辑(最常用命令之一,可按指定列编辑)
mutate 以正则表达式基于已有列创建新的一列(常用于生成多列测试数据)
mutate2 用类似awk的数值/表达式,以正则表达式基于已有(多)列创建新的一列(常用)
gather 类似于R里面tidyr包的gather方法# 排序
sort 按指定(多)列进行排序# 绘图
plot 基本绘图
plot hist histogram 直方图
plot box boxplot 箱线图
plot line line plot and scatter plot 线图和散点图# 其它
version 版本信息和检查新版本
genautocomplete 生成支持Bash自动补全的配置文件,重启Terminal生效。
使用
输入数据要求每行的列数一致,空行也会报错
csvtk默认输入数据含有标题行,如没有请开启全局参数-H
csvtk默认输入数据为CSV格式,如为TSV请开启全局参数-t
输入数据列名最好唯一无重复
如果TSV中存在双引号"",请开启全局参数-l
csvtk默认以#开始的为注释行,若标题行含#,请给全局参数-C指定另一个不常见的字符(如$)
本次使用的数据是扩增子的otu表格和注释表格,还有分组文件。
# 下载测试数据
for i in otutab.txt taxonomy.txt metadata.txt otus.fa;dowget -c http://210.75.224.110/github/MicrobiomeStatPlot/Data/Science2019/$i; done
mv metadata.txt metadata.tsv
# 转换TSV为CSV
sed 's/\t/,/g' metadata.tsv > metadata.csv
# 删除行名的#号,旧版本格式,目前不常用
sed -i '1 s/#//' otutab.txt
csvtk pretty让打印出来的内容排版更加美丽,其实对于tsv文件影响不大,要加上全局参数-t,但是对于csv文件影响还是挺大的。
注意csvtk主要用于处理csv数据,对于txt数据尽量转化为csv数据整理,否则好多操作都不能实现。
# 预览csv
head -n3 metadata.csv# SampleID,Group,Date,Site,Sequencing,Platform,Species,Batch,BarcodeSequence,LinkerPrimerSequence,ReversePrimer
# KO1,KO,2017/6/30,Beijing,BGI,HiSeq2500,Arabidopsis,1,ACGCTCGACA,AACMGGATTAGATACCCKG,ACGTCATCCCCACCTTCC
# KO2,KO,2017/6/30,Beijing,BGI,HiSeq2500,Arabidopsis,1,ATCAGACACG,AACMGGATTAGATACCCKG,ACGTCATCCCCACCTTCC# 可视化csv
csvtk pretty metadata.csv|head -n3# SampleID Group Date Site Sequencing Platform Species Batch BarcodeSequence LinkerPrimerSequence ReversePrimer
# KO1 KO 2017/6/30 Beijing BGI HiSeq2500 Arabidopsis 1 ACGCTCGACA AACMGGATTAGATACCCKG ACGTCATCCCCACCTTCC
# KO2 KO 2017/6/30 Beijing BGI HiSeq2500 Arabidopsis 1 ATCAGACACG AACMGGATTAGATACCCKG ACGTCATCCCCACCTTCC# TSV格式使用pretty 打印样式影响不大
head -n3 otutab.txt
# 自动去除了表头
csvtk -H pretty otutab.txt | head -n3
注意csv格式正常统计,但是tsv格式统计列数量错误;
csvtk stat metadata.csv
# file num_cols num_rows
# metadata.csv 11 18#--注意使用tsv格式文件需要添加参数 -t
csvtk stat metadata.tsv -t
# file num_cols num_rows
# metadata.tsv 1 18
-h, --help help 文件调用-v, --verbose 打印冗余信息,用#号分隔的文件名称
# 默认去除#号注释行,如果存在#号行,-C指定一个不常用字符
csvtk -t headers otutab.txt -C !
#OTUID
#KO1
#KO2
···csvtk headers otutab.txt -t -v
# otutab.txt
#1 #OTUID
#2 KO1
#3 KO2
···
这里需要注意的式-n参数可以用于多文件统计,没有表头记得加上-H参数。
--cols 只打印文件列数量-h, --help 调用帮助文件-n, --no-files 不打印文件名--rows 只打印文件行数量--tabular 输出机器友好的tab格式文件
# 默认输出列和行数量
csvtk -t dim otutab.txt
#file num_cols num_rows
#otutab.txt 19 2,631#仅仅输出行数量
csvtk -t nrow otutab.txt
#2631# 仅仅输出列数量
csvtk ncol otutab.txt -t
# 19#--注意同时统计多个文件可以在后面加上-n参数csvtk dim *.txt -t -n#file num_cols num_rows
#otutab.txt 19 2,631
#taxonomy.txt 8 2,631
选定的数字或文本字段的汇总统计信息(按组分组字段),注意不能混用列名和数字标识。
所支持的函数:countn (count numeric values), min, max, sum,
mean, stdev, variance, median, q1, q2, q3,
entropy (Shannon entropy),
prod (product of the elements);
支持的调用语法:
count, first, last, rand, unique, collapse, countunique。
Flags:-n, --decimal-width int 限制浮点数为N个小数点(默认为2)-f, --fields strings 统计类型指定:operations on these fields. e.g -f 1:count,1:sum or -f colA:mean. available operations: collapse, count, countn, countunique, entropy, first, last, max, mean, median, min, prod, q1, q2, q3, rand, stdev, sum, uniq, variance-g, --groups string 分组,按照行或者列:group via fields. e.g -f 1,2 or -f columnA,columnB-h, --help 调用帮助文件-i, --ignore-non-numbers 忽略NA值:"NA" or "N/A"-S, --rand-seed int 设定随机种子,默认11 "rand" (default 11)-s, --separater string separater for collapsed data (default "; ")
下面就一个列进行统计,主要是训练这些常用函数的用法。很多时候可以联合使用。注意经常我们的数据中有NA存在,此时加上-i。
#-对指定列WT2进行求和,获得某样本的总数据量
cat otutab.txt | csvtk summary -f WT2:sum -t
# 求均值
cat otutab.txt | csvtk summary -f WT2:mean -t
# 统计该列的长度,即行数
cat otutab.txt | csvtk summary -f WT2:countn -t#-对应的max,min和first,last是一个道理,也就是下面的命令同等有效:
cat otutab.txt | csvtk summary -f WT2:first -t
cat otutab.txt | csvtk summary -f WT2:max -t# 统计方差:variance
cat otutab.txt | csvtk summary -f WT2:variance -t# 统计标准差
cat otutab.txt | csvtk summary -f WT2:stdev -t# 提取非重复值
cat otutab.txt | csvtk summary -f WT2:uniq -t
# 对去除重复值后的值统计数量
cat otutab.txt | csvtk summary -f WT2:countunique -t
对多个列同时进行统计:
cat otutab.txt | csvtk summary -f WT2:countunique,OE1:uniq -t
cat otutab.txt | csvtk summary -f WT2:mean,OE1:mean -t
该功能和R包dplyr的许多功能类似,用法类似,例如这个功能:共有连接,左连接,全部连接。可以支持多个共有列。默认如果不加参数,会进行共有列连接。
csvtk join -h-f, --fields string Semicolon separated key fields of all files, if given one, we think all the files have the same key columns. Fields of different files should be separated by ";", e.g -f "1;2" or -f "A,B;C,D" or -f id (default "1")-F, --fuzzy-fields using fuzzy fields, e.g., -F -f "*name" or -F -f "id123*"-h, --help help for join-i, --ignore-case ignore case-k, --keep-unmatched keep unmatched data of the first file (left join)-L, --left-join left join, equals to -k/--keep-unmatched, exclusive with --outer-join--na string content for filling NA data-O, --outer-join outer join, exclusive with --left-join
实例,这里我们的实例都是来自于微生物组分析得到的表格,让大家可以更好的应用于微生物组数据分析和实践。
#-合并OTU表格和注释文件 -f 1 指定第一列为合并公共列
csvtk -t join -f 1 otutab.txt taxonomy.txt | csvtk -t dim
#file num_cols num_rows
#- 26 2,631
# 保存结果,供后面使用
csvtk -t join -f 1 otutab.txt taxonomy.txt > otutax.txt# 演示一下左连接:仅保留第一个文件的列
head otutab.txt > otutab_sub.txt
csvtk -t join -f 1 otutab_sub.txt taxonomy.txt --left-join | csvtk -t dim
#file num_cols num_rows
#- 26 9#--如果左连接第二个表格没有对应的信息,则使用NA填充空位
head -n9 taxonomy.txt > taxonomy_sub.txt
csvtk -t join -f 1 otutab_sub.txt taxonomy_sub.txt --na NA --left-join
#--全部合并,对于空缺位置可以随意标示,0,或者NA或者自己的名字也可以
csvtk -t join -f 1 otutab_sub.txt taxonomy_sub.txt --outer-join --na wentao
# 通过列名来指定合并列
csvtk -t join -f "OTUID;OTUID" otutab.txt taxonomy.txt |csvtk -t dim
# 准备一下无表头的表
cut -f 1-3 metadata.tsv | tail -n+2 | head -n2 > metatest.txt
cat metatest.txt
#KO1 KO 2017/6/30
#KO2 KO 2017/6/30# 添加表头
csvtk -t add-header -n SampleID,Group,Date metatest.txt
#SampleID Group Date
#KO1 KO 2017/6/30
#KO2 KO 2017/6/30
当我知道csvtk和seqkit是一个作者写的后,我就开始肆无忌惮的的联用这两个工具了,seqkit的安装和使用参考如下教程:
seqkit:序列梳理神器-统计、格式转换、长度筛选、质量值转换、翻译、反向互补、抽样、去重、滑窗、拆分等30项全能
例如下面我们将演示三个表格合并,此时需要处理一下代表序列文件,作为除了otu表格和注释表格之外的第三个表格:将fa格式转化为tsv格式,删除结尾多余制表符,再添加表头
seqkit fx2tab otus.fa|cut -f 1-2 |csvtk -t add-header -n OTUID,Sequence > otus_fa.txt
# 然后转化为csv格式预览,按q退出预览
csvtk tab2csv otus_fa.txt|less -S
# 通过第一列合并
csvtk -t join -f 1 otutab.txt taxonomy.txt otus_fa.txt |head
# 通过共有列名合并
csvtk -t join -f OTUID otutab.txt taxonomy.txt otus_fa.txt |head
# -g 参数指定分组
cat otutax.txt | csvtk -t summary -i -f OE2:mean,WT2:mean -g Phylum
# 指定多个分组
cat otutax.txt | csvtk -t summary -i -f OE2:mean,WT2:mean -g Phylum,Class
转置这一个步骤相当消耗时间,尤其是宏基因组的较大的表格。
#--转置,提取表头,打印前面几个
csvtk -t transpose otutab.txt |csvtk -t headers |head
# -f 参数可以使用数字指定列 并挑选出来
csvtk -t cut -f 1,3 otutab.txt | csvtk -t headers
# -f 也可以通过列名来指定并打印列
csvtk -t cut -f OE2 otutab.txt | csvtk -t headers
# 去掉列只需要在序号前面加上-即可,这里去除第二和第三列
csvtk -t cut -f -2,-3 otutab.txt | csvtk -t headers
# 指定范围去除某些列,这里去除第二列到第六列
csvtk -t cut -f -6--2 otutab.txt | csvtk -t headers
# 注意负号添加在列名字前面同样有效
csvtk -t cut -f -OE1 otutab.txt | csvtk -t headers
使用-F参数,使用双引号,可以使用正则而表达式。
# 筛选W和OE开头的列
csvtk -t cut -F -f "OTUID,W*,OE*" otutab.txt | csvtk -t headers
#--无论是按照数字还是列名指定列,这个顺序就是打印出来的顺序。
csvtk -t cut -F -f "OTUID,OE1,WT1" otutab.txt | csvtk -t headers
在用markdown写笔记,或做网页时,经常需要把表格排版为markdown格式,手动排版非常耗时。使用此工具会极方便
cut -f 1-7 metadata.tsv | csvtk -t csv2md
SampleID|Group|Date |Site |CRA |CRR |BarcodeSequence
:-------|:----|:--------|:--------|:---------|:--------|:--------------
KO1 |KO |2017/6/30|Chaoyang |CRA002352 |CRR117575|ACGCTCGACA
KO2 |KO |2017/6/30|Chaoyang |CRA002352 |CRR117576|ATCAGACACG
KO3 |KO |2017/7/2 |Changping|CRA002352 |CRR117577|ATATCGCGAG
# 无需指定输出,会生成 otutab.txt.xlsx 的Excel表格输出
csvtk -t csv2xlsx otutab.txt
# -o 指定输出文件名
csvtk -t csv2xlsx otutab.txt -o otutab.xlsx
Flags:-h, --help help for xlsx2csv-a, --list-sheets 转化全部的sheet-i, --sheet-index int 指定sheet (default 1)-n, --sheet-name string sheet to retrieve
# -将xlsx转化为csv文件,显示表格中sheet编号
csvtk xlsx2csv -a otutab.txt.xlsx# 转化第一张sheet,输出到屏幕
csvtk -t xlsx2csv -i 1 otutab.txt.xlsx | head
-n, --number int 指定需要查看数据的行数
# 查看前十行数据
csvtk head otutab.txt -n 10
Flags:-h, --help help for concat-i, --ignore-case 忽略列名 (column name)-k, --keep-unmatched keep blanks even if no any data of a file matches-u, --unmatched-repl string replacement for unmatched data
# 需要列名一样, 但是顺序可以不一样,并去除多余表头
wc -l otutab_sub.txt otutab.txt
csvtk -t concat otutab_sub.txt otutab.txt | wc -l
# 不同时默认共有列合并,-i忽略大小写
csvtk -t concat otutab_sub.txt otutab.txt -i | csvtk -t stat
# -u 将未匹配上的行使用Unmached补全
csvtk concat otutab_sub.txt otutab.txt -u Unmached | csvtk -t stat
Flags:-h, --help help for sample-n, --line-number 打印第一列,行号 ("n")-p, --proportion float 按照比例抽样-s, --rand-seed int 随机种子(default 11)
# 按照比例抽样,抽取一半,没有表头添加参数-H
seq 100 | csvtk sample -H -p 0.5 | wc -l
#46# 抽取十分之一
seq 100 | csvtk sample -H -p 0.1 | wc -l
# 10
# 打印行号
seq 100 | csvtk sample -H -p 0.05 -n
#50,50
#52,52
#65,65
例子:1. 选择单个列,根际列序号或者列名csvtk cut -f 1csvtk cut -f colA2. 多个列选择 (可以重复列名,调整顺序)csvtk cut -f 1,3,2,1csvtk cut -f colA,colB,colA3. 选择列可用于列排序csvtk cut -f 1,3-5 # 1, 3, 4, 5csvtk cut -f 3,5- # 3rd col, and 5th col to the end 第五列发放最后面csvtk cut -f 1- # 全选列csvtk cut -f 2-,1 # move 1th col to the end4. 负号放到前面代表去除某一列csvtk cut -f -1,-3 # discard 1st and 3rd columncsvtk cut -f -1--3 # discard 1st to 3rd columncsvtk cut -f -2- # discard 2nd and all columns on the right.csvtu cut -f -colA,-colB # discard colA and colBFlags:-f, --fields string 选择列. type "csvtk cut -h" for examples-F, --fuzzy-fields 模糊选择,正则表达书,用单引号括起来, e.g., -F -f "*name" or -F -f "id123*"-h, --help help for cut-i, --ignore-case ignore case (column name)-u, --uniq-column deduplicate columns matched by multiple fuzzy column names
# 列名选择
csvtk -t cut -f OE2 otutab.txt |head
#OE2
#1610
#497# 序号选择
csvtk -t cut -f 2,3,5 otutab.txt |head
#KO1 KO2 KO4
#1073 1926 1356
#1965 1233 2241# 模糊选择
csvtk -t cut -F -f 'OE*' otutab.txt |head
#OE1 OE2 OE3 OE4 OE5 OE6
#1259 1610 1337 944 1245 1013
#641 497 1225 1271 948 638# 去除某些列
csvtk -t cut -f -1,-2,-3 otutab.txt |head
#KO3 KO4 KO5 KO6 OE1 OE2
#810 1356 1064 1069 1259 1610
#2368 2241 2901 1835 641 497 # 选择第八列以及之后的全部列
csvtk -t cut -f 8- otutab.txt |head
#OE1 OE2 OE3 OE4 OE5 OE6 WT1 WT2 WT3 WT4 WT5 WT6
#1259 1610 1337 944 1245 1013 2303 2512 1698 1974 1441 1544
#641 497 1225 1271 948 638 1286 1499 843 1122 1496 1177# 选择第八列到第十列
csvtk -t cut -f 8-10 otutab.txt |head
#OE1 OE2 OE3
#259 1610 1337
按照某列去除重复
# 挑选界,对界进行去除重复
csvtk -t cut -f 2 taxonomy.txt |csvtk uniq -f 1
#Kingdom
#Bacteria
#Archaea# 直接对注释文件某列去除重复,不考虑其他列
csvtk -t uniq -t -f 2 taxonomy.txt
#OTUID,Kingdom,Phylum,Class,Order,Family,Genus,Species
#ASV_657,Bacteria,Actinobacteria,Actinobacteria,Actinomycetales,Thermomonosporaceae,Unassigned,Unassigned
#ASV_1646,Archaea,Thaumarchaeota,Unassigned,Nitrososphaerales,Nitrososphaeraceae,Nitrososphaera,Unassigned
Flags:-f, --fields string select only these fields. e.g -f 1,2 or -f columnA,columnB (default "1")-F, --fuzzy-fields 模糊选择, e.g., -F -f "*name" or -F -f "id123*"-i, --ignore-case ignore case-r, --reverse 反转排序-n, --sort-by-freq 按照频数排序-k, --sort-by-key 按照键排序
# 对第二列统计频数
csvtk -t freq -f 2 taxonomy.txt
#Kingdom,frequency
#Archaea,1
#Bacteria,2630# 按数值反转排序
csvtk -t freq -f 2 taxonomy.txt -n -r
#Kingdom,frequency
#Bacteria,2630
#Archaea,1
csvtk -t inter otutab.txt taxonomy.txt |head
#OTUID
#ASV_2700
#ASV_2092
Flags:-f, --fields string comma separated key fields, column name or index. e.g. -f 1-3 or -f id,id2 or -F -f "group*" (default "1")-F, --fuzzy-fields 模糊选择, e.g., -F -f "*name" or -F -f "id123*"-h, --help help for grep-i, --ignore-case ignore case-v, --invert 去除匹配上的行-n, --line-number 打印行号 ("n")-N, --no-highlight 不高亮-p, --pattern strings 优雅答应选项(multiple values supported)-P, --pattern-file string pattern files (one pattern per line)
# 选择第3列门是Actinobacteria 的行
csvtk -t grep -f 3 -p Actinobacteria taxonomy.txt # 可用列表 -P 接匹配列表
# 构建一个id列表,并去除表头
csvtk -t cut -f 1 otutab.txt | head | csvtk del-header > id.txt
# 使用-P选项后面接列表,比awk筛选更方便
csvtk -t grep -f 1 -P id.txt taxonomy.txt
# 选择OE2 列序列数量大于500的行
csvtk -t filter -f "OE2>500" otutab.txt#支持多列共同筛选
csvtk -t filter -f "2-5>300" otutab.txt# 注意模糊匹配使用-F选项,所有列均满足条件
csvtk -t filter -F -f "OE*>500" otutab.txt
#OTUID,KO1,KO2,KO3,KO4,KO5,KO6,OE1,OE2,OE3,OE4,OE5,OE6,WT1,WT2,WT3,WT4,WT5,WT6
#ASV_657,1073,1926,810,1356,1064,1069,1259,1610,1337,944,1245,1013,2303,2512,1698,1974,1441,1544
# 按照门进行拆分文件-会以门的名字命名
csvtk -t split taxonomy.txt -f Phylum
# 查看分出来的文件
ls taxonomy-*
# 删除这些文件,太多使目录变混乱
rm taxonomy-*# 通过指定-o输出到一个文件夹中
mkdir -p split
csvtk -t split taxonomy.txt -f Phylum -o split/# 指定两个列进行分析
csvtk -t split taxonomy.txt -f Phylum,Class -o split/
# 两两组合
csvtk -t cut -f 1,2,3 taxonomy.txt |head -n 2 |csvtk -t comb -n 2
#ASV_657,Bacteria
#ASV_657,Actinobacteria
#Bacteria,Actinobacteria# 三个组合
csvtk -t cut -f 1,2,3,4 taxonomy.txt |head -n 2 |csvtk -t comb -n 3# 断棍模型-单个到全部组合都做出来
csvtk -t cut -f 1,2,3,4 taxonomy.txt |head -n 2 |csvtk -t comb -n 0
# seq生成序列,mutate添加一列,add-header--添加列名为a,b
seq 3 | csvtk mutate -H |csvtk add-header -n a,b
#a,b
#1,1
#2,2# 可以分别用-n参数指定-n a -n b
seq 3 | csvtk mutate -H csvtk add-header -n a -n b
#a,b
#1,1
#2,2
# 添加列名
seq 3 | csvtk add-header
#添加后删除列名
seq 3 | csvtk add-header | csvtk del-header# 或者使用-H参数指定无表头防止误删
seq 3 | csvtk del-header -H
csvtk -t cut -f 1-2 taxonomy.txt |head |csvtk -t rename -f 1-2 -n B,P
#B,P
#ASV_657,Bacteria
#ASV_2,Bacteria
cut -f1-3 otutab.txt |csvtk -t round -n 2 -f KO1 |head
#OTUID KO1 KO2
#ASV_657 1073.00 1926
#ASV_2 1965.00 1233
# 使用已有列改名 作为新的一列
csvtk -t mutate -f Class -n Class2 taxonomy.txt | head -n3
#OTUID Kingdom Phylum Class Order Family Genus Species Class2
#ASV_657 Bacteria Actinobacteria Actinobacteria Actinomycetales Unassigned Unassigned Unassigned Actinobacteria
#ASV_2 Bacteria Proteobacteria Betaproteobacteria Burkholderiales Comamonadaceae Pelomonas Pelomonas_puraquae Betaproteobacteria# 复杂一点:将otu表格otu名字种otu部分 提取出来做为group列
csvtk -t mutate -f 1 -n group -p "^(.+?)[0-9]" otutab.txt |head -n3
#OTUID KO1 KO2 KO3 KO4 KO5 KO6 OE1 OE2 OE3 OE4 OE5 OE6 WT1 WT2 WT3 WT4 WT5 WT6 group
#ASV_657 1073 1926 810 1356 1064 1069 1259 1610 1337 944 1245 1013 2303 2512 1698 1974 1441 1544 ASV_
#ASV_2 1965 1233 2368 2241 2901 1835 641 497 1225 1271 948 638 1286 1499 843 1122 1496 1177 ASV_
# 使用字符串重复,作为一个新列
cut -f 1-3 otutab.txt |csvtk -t mutate2 -t -e " 'abc' " -n group |head -n3
#OTUID KO1 KO2 group
#ASV_657 1073 1926 abc
#ASV_2 1965 1233 abc# 列 合并,如果是字符串,可以合并在一起
cut -f 1-4 taxonomy.txt |csvtk -t mutate2 -n Comname -e ' $Class + "-" + $Phylum ' |head
#OTUID Kingdom Phylum Class Comname
#ASV_657 Bacteria Actinobacteria Actinobacteria Actinobacteria-Actinobacteria
#ASV_2 Bacteria Proteobacteria Betaproteobacteria Betaproteobacteria-Proteobacteria
#ASV_3 Bacteria Proteobacteria Gammaproteobacteria Gammaproteobacteria-Proteobacteria#如果是数值可以进行基本算数运算-例如两列求和
cut -f1-7 otutab.txt |csvtk -t mutate2 -n sum12 -e ' $3 + $2 ' |head
#OTUID KO1 KO2 KO3 KO4 KO5 KO6 sum12
#ASV_657 1073 1926 810 1356 1064 1069 2999.00
#ASV_2 1965 1233 2368 2241 2901 1835 3198.00# 使用逻辑判断,添加新列
cut -f1-7 otutab.txt | csvtk -t mutate2 -n sum12 -e ' $2>1000' |head
#OTUID KO1 KO2 KO3 KO4 KO5 KO6 sum12
#ASV_657 1073 1926 810 1356 1064 1069 true
#ASV_2 1965 1233 2368 2241 2901 1835 true
#ASV_3 567 460 898 902 1224 854 false# 可以使用条件分配逻辑判断的填充
cut -f1-7 otutab.txt |csvtk -t mutate2 -n sum12 -e ' $3>1000? "small" : "big"' |head
#OTUID KO1 KO2 KO3 KO4 KO5 KO6 sum12
#ASV_657 1073 1926 810 1356 1064 1069 small
#ASV_2 1965 1233 2368 2241 2901 1835 small
#ASV_3 567 460 898 902 1224 854 big
# 想知道每个门中有哪些ASV
cat taxonomy.txt |csvtk -t collapse -f 1,2,3 -v 1 -s ';' | less -S > phylum_ASV.txt
# -n 指定分隔出来的内容名称,这里指定两个,那就只会分出来两个,多指定几个,那就多分出来几个。
csvtk -t sep phylum_ASV.txt -f3 -s ";" -n OTU1,OTU2 --drop | less -S
#Kingdom Phylum OTUID OTU1 OTU2
#Bacteria Ignavibacteriae ASV_1159;ASV_2278 ASV_1159 ASV_2278
#Archaea Thaumarchaeota ASV_1646 ASV_1646
#Bacteria Armatimonadetes ASV_989;ASV_1709;ASV_1839;ASV_2014 ASV_989 ASV_1709
#Bacteria Gemmatimonadetes ASV_2825 ASV_2825# --merge 会将剩下的内容放到最后一列
csvtk -t sep phylum_ASV.txt -f3 -s ";" -n OTU1,OTU2 --merge| less -S
# 按照第一列作为索引,将数据变为长数据,方便出图
cat otutab.txt |csvtk -t gather -k SampleID -v value -f -1 |head
#OTUID SampleID value
#ASV_657 KO1 1073
#ASV_657 KO2 1926
#ASV_657 KO3 810# 下面使用id和界作为索引,变化为长数据
cat otutax.txt |csvtk -t gather -k id -v value -f -1,-21 |head
#OTUID Phylum id value
#ASV_657 Actinobacteria KO1 1073
#ASV_657 Actinobacteria KO2 1926
# 选择门和科这两个等级数据,并去除重复,然后按照门,将科归类到 门后面,默认使用 “;” 分隔
# 某个门中有哪 些科,和collapse有点类似,按; 分隔
cat taxonomy.txt | csvtk -t uniq -f Phylum,Class |csvtk -t fold -f Phylum -v Class# 换用逗号和空格作为分隔符
cat taxonomy.txt | csvtk -t uniq -f Phylum,Class |csvtk -t fold -f Phylum -v Class -s ", "# 下面按照门和钢给OTU做分类
cat taxonomy.txt |csvtk -t collapse -f 1,2,3 -v 1 -s ';' | head
由于使用go出图并不能达到我们的发文预期,所以这里只是简单的看看。优点是命令行一行完成,不用像R解决文件读写,更高效。
# 对表格的第三列绘制直方图
csvtk -t plot hist otutab.txt -f 3 --title Histogram -o histogram.png
# 直接展示
csvtk plot hist otutab.txt -f 3 --title Histogram | display
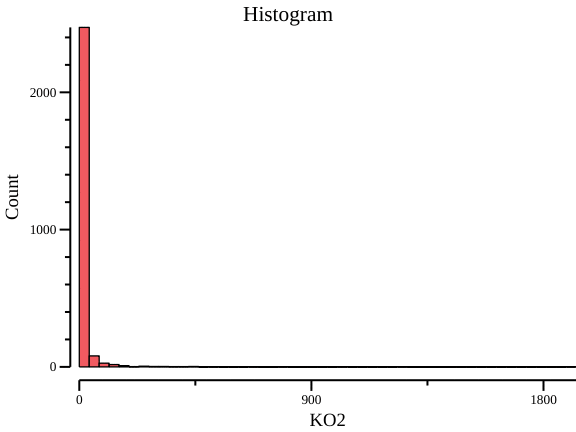
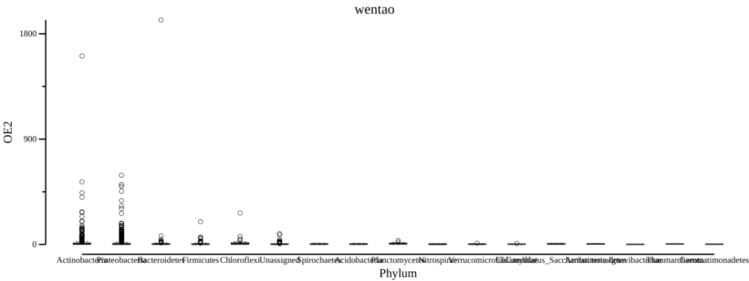
# 设置-g参数用于指定x周坐标,-f 指定y周内容
csvtk -t plot box otutax.txt -g Phylum -f "OE2" --width 12 --title "wentao" > boxplot.png
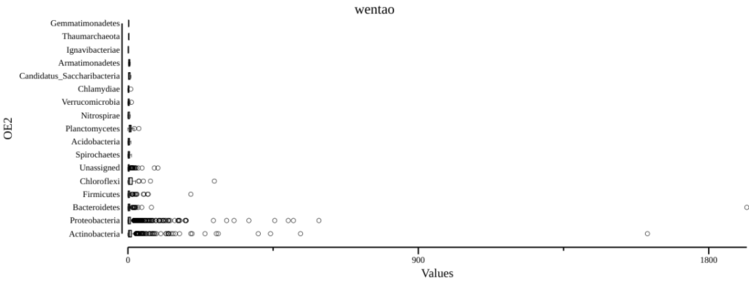
# 坐标轴反转一下
csvtk -t plot box otutax.txt -g Phylum -f "OE2" --width 12 --horiz --title "wentao" > boxplot2.png
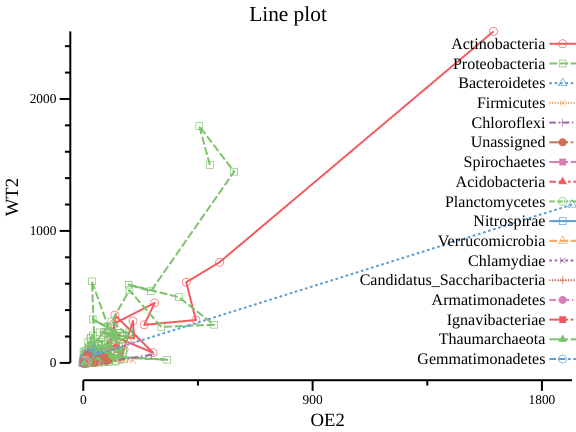
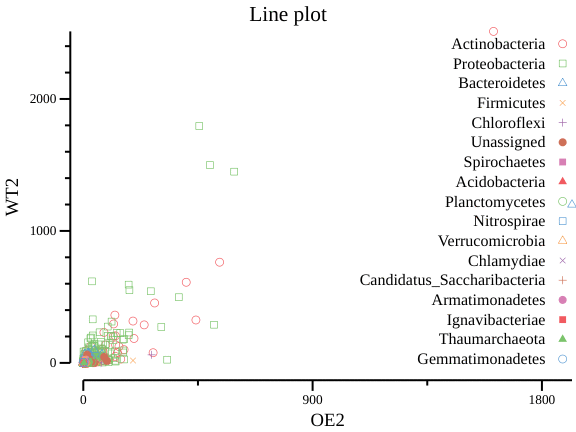
csvtk -t plot line otutax.txt -x OE2 -y WT2 -g Phylum --title "Line plot" > lineplot.pngcsvtk -t plot line otutax.txt -x OE2 -y WT2 -g Phylum --title "Line plot" --scatter > lineplot2.png
csvtk -t watch -O hist.pdf -f WT1 otutab.txt
这里使用的txt文件,所以添加了-t全局参数,这里作者帮助文件显示只能计算这一种相关,如果我们做微生物组分析,斯皮尔曼相关更加适合
csvtk -t corr -i -f WT1,WT2 otutab.txt
# WT1 WT2 0.9373
head -n3 otutab.txt | csvtk -t csv2json
功能更强大,但用法稍微不同。
csvtk -t filter2 -f '$OE1 > 500 || $WT2 > 100' otutab.txt |head
#OTUID,KO1,KO2,KO3,KO4,KO5,KO6,OE1,OE2,OE3,OE4,OE5,OE6,WT1,WT2,WT3,WT4,WT5,WT6
#ASV_657,1073,1926,810,1356,1064,1069,1259,1610,1337,944,1245,1013,2303,2512,1698,1974,1441,1544
#ASV_2,1965,1233,2368,2241,2901,1835,641,497,1225,1271,948,638,1286,1499,843,1122,1496,1177
csvtk xlsx2csv -a otutab.txt.xlsx
软件管方教程:https://bioinf.shenwei.me/csvtk/usage
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读






 京公网安备 11010802041100号 | 京ICP备19059560号-4 | PHP1.CN 第一PHP社区 版权所有
京公网安备 11010802041100号 | 京ICP备19059560号-4 | PHP1.CN 第一PHP社区 版权所有