Corset: enabling differential gene expression analysis for de novo assembled transcriptomes
背景:
转录组测序这种高通量RNA测序,是一个非常强力的技术
去研究转录本的各个方面
it has a broad range of applications 它有着广泛的应用
包括发现新的基因,检测可变剪接,差异表达基因,基因融合检测,比如SNPs和转录后的编辑post-
transcriptional editing [1,2] 变异鉴定,
One of the advantages of
RNA-seq over older technology, such as microarrays,
is that it enables the transcriptome-wide analysis of
non-model organisms because a reference genome and
annotation are not required for generating and analyzing
the data.
When no reference genome is available, the tran-
scriptome is de novo assembled directly from RNA-seq
reads [3]
Several programs exist for de novo transcriptome
assembly: Oases [4] and Trans-abyss [5], which extend the
Velvet [6] and Abyss [7] genomic assemblers,
, respectively,
as well as purpose built transcriptome assemblers such
as Trinity [8]. These programs are capable of assembling
millions of short reads into transcript sequences - called
contigs.
One common and biologically important application
of RNA-seq is identifying genes that are differentially
expressed between two or more conditions [9].
找差异表达基因
However,
performing a differential expression analysis on a de novo
assembled transcriptome is challenging because multiple
contigs per gene are reported. Multiple contigs, with
shared sequence, arise because transcriptome assemblers
differentiate between isoforms of the same gene, and
report each separately.
然而,从头组装转录本寻找差异表达基因面临挑战,因为一个基因包含多个contigs
很多的contigs都有共享reads......
Furthermore, they often report
contigs that are not truly representative of different
isoforms but arise from artifacts such as sequencing
errors, repeats, variation in coverage or genetic variation
within a diploid individual or pooled population.
此外,常说contigs不能真正代表不同的可变剪接,由于人为因素引起的测序错误
重复,覆盖度的变化和基因突变,在二倍体...
As aresult, transcriptome assemblers often report fragmented
versions of a transcript, or repeated contigs that differ only
by a SNP or indel.
Surprisingly, simulations have shown
that even the assembly of data without any sequencing
errors, SNPs or alternative splicing can generate multiple
contigs per gene [10].
令人意外的,模拟测试结果表明尽管组装的数据没有任何测序错误,SNPS
或者可变剪接还是能对每个基因产生大量的contigs
Hence, the number of contigs
produced by a de novo assembly is typically large; for
example, assemblies with 80 million reads can produce
hundreds of thousands of contigs [11].
因此从头组装产生的contigs得数量还是很大,比如,8000千万的reads几十万的contigs
The inevitably long list of contigs generated by de novo
transcriptome assembly causes several issues for differential
expression analysis:
从头组装转录本不可避免地产生一连串contigs 在差异表达部分产生了很多问题:
i) reads cannot be aligned unambiguously to duplicated sequences and determining the origin
of ambiguously aligned reads is error prone;
reads 不能准确地比对到重复序列和界定原始模糊比对的reads,这是容易出错的。
ii) the statistical power of the test for differential expression is reduced
as reads must be allocated amongst a greater number of
contigs, thus reducing the average counts per contig;
差异表达的统计结果是减少的,reads 必须分配到很多的contigs中,正如减少了每个contigs的counts
iii),
the adjustment for multiple testing is more severe;
对多次测试的调整是更加严格的
and iv),
once differentially expressed contigs have been identified,
interpretation is difficult, as many genes will be present in
the list multiple times.
一旦差异表达的contigs被鉴定出来,解释是困难的,因为很多基因将会存在于列表多次。
Performing a differential expression
analysis on genes, rather than contigs, would overcome these difficulties.
对基因进行差异表达分析,而不是contigs,必须克服这些困难
However, the procedure for estimating
gene-level expression from a set of de novo assembled
contigs is not straightforward and has not been thoroughly
examined in the literature.
然而,从一系列的从头组装的contigs 中评估基因水平的表达不是很直接的,而且到现在还没有在文献中被完全检验出来
#========================================
Several steps are involved in identifying differentially
expressed genes from a de novo assembled transcriptome
(Figure 1): RNA-seq reads are first assembled, reads are
next mapped back to contigs, contigs are then clustered
into genes, after which the expression level for each gene
cluster is summarized, and statistical testing is performed
to detect differential expression.
从从头组装鉴定差异基因的表达涉及到很多的步骤,
转录组序列先是组装,然后回帖到contigs,contig在聚成基因,
当每个基因的差异表达水平被总结后,就进行差异表达基因的统计测试

Several studies have compared individual steps in this
analysis pipeline. For example, the relative merits of
different de novo assemblers and steps prior to assembly,
有不少研究比较这个分析流程的每一步,比如不同的从头组装软件跟先前组装的方法的相对有点
dashed box:虚线框
such as quality control, have been examined [12–15].
比如质控,在引用文献中的12-15中已经被比较过了
Similarly, the choice of method for performing count-based
statistical testing for differential expression has been
evaluated [16,17].
同样的,基于count的的差异表达统计方法也被评估过了
However, few studies have compared
or even suggested a path for obtaining gene-level counts
from transcriptome assemblies [18,19]
然而,很少有研究比较或则建议某种方法从转录本组装去获得基因水平的counts
18. Sandmann T, Vogg MC, Owlarn S, Boutros M, Bartscherer K: The head-
regeneration transcriptome of the planarian Schmidtea mediterranea.
Genome Biol 2011, 12:R76.
19. Oono Y, Kobayashi F, Kawahara Y, Yazawa T, Handa H, Itoh T,
Matsumoto T: Characterisation of the wheat (triticum aestivum L.)
transcriptome by de novo assembly for the discovery of phosphatestarvation-responsive genes: gene expression in Pi-stressed wheat.
BMC Genomics 2013, 14:77.
and only a single
automated pipeline has thus far (起劲)been implemented to
address this need(解决这一需求) [20];
仅有一个自动化的流程迄今已实现解决这一需求
it is provided by Trinity to run
RSEM [21] followed by edgeR [22] or DESeq [23].
它有trinity组装后用RSEM 定量后用edgeR或DESeq做差异
This pipeline is inflexible, however, to the choice of assembler.
然而 这个流程对组装软件的选择是不灵活的
In this paper we present Corset, a method and software
for obtaining gene-level counts from any de novo tran-
scriptome assembly
在这个而文章中,我们提出corset,一个方法和软件从从头组装的转录本组装去获得基因水平的counts
Corset takes a set of reads that have
been multi-mapped (multiple alignments per read are
reported) to the de novo assembled transcriptome and
hierarchically clusters the contigs based on the proportion
of shared reads and expression patterns.
corset需要一套在无参组装中有比对到多个地方的reads 和 基于共享reads的比例和表达模式进行分层次的聚类contigs
Expression
patterns allow for discrimination(区别) between genes that
share sequence, such as paralogues, if the expression
levels between groups are different.
(paralogues:旁系同源的)
如果基因的表达水平在不同组是不同的,表达模式允许基因间共享reads之间诸如旁系同源的区别。
Using the mapped
reads, Corset then outputs gene-level counts. The gene-
level counts can then easily be tested for differential
expression using count-based frameworks such as edgeR
and DESeq.
corset利用比对上的reads输出基因水平的counts,基因水平的counts能容易地用于基于counts流程的差异表达方法
We demonstrate that Corset consistently
performs well compared to alternative clustering methods
on a range of metrics.
我们论证 corset 比其他可选择的聚类方法在很多方面都好
Moreover, as it is an assembler-
independent method, it allows contigs and transcripts
from various sources to be combined. It is also simpler to
use, with the clustering and counting steps encompassed(包含)
in a single run of the software.
此外,因为它作为一个独立的方法,它允许来源不同的contigs或转录组合在一起
它集聚类和计算表达量与一起,也是一个更简单实用的软件
Results and discussion
结果和讨论
Corset clusters contigs and counts reads
corset对contigs和计算表达量
The first step in performing a gene-level differential ex-
pression analysis for a non-model organism is to assemble
the contigs, which can be performed using a variety of
software.
没有参考基因的物种第一步去算基因差异表分析是先组装contigs,这一步可以用很多软件来执行
As previously outlined, (概述)
this process produces
multiple sequences or contigs per gene. Consequently, the
next step is to group, or cluster, the contigs into genes to
facilitate (促进)downstream differential expression analysis. This
clustering step is the first step of Corset
正如之前的概述,这个流程美格基因产生大量的序列或则contigs,因此下一步是成组或则成簇,将contigs成组或则成簇成基因,
促进下游差异表达的分析,这一步聚类是corset的第一步
Corset requires that, after transcriptome assembly, reads
are mapped back to the contigs allowing reads to map to
multiple contigs (multi-mapping).
在组装后,reads回帖到contigs,corset需要比对到多个位置的contigs
These multi-mapped
reads are then used as a proxy for detecting sequence
similarity between contigs, as well as providing informa-
tion about the expression level of the contigs.
这些比对到多个位置的reads被当成代理去检测contigs之间的相似度,并且提供信息contigs水平的差异表达信息
Corset also
uses the read information to filter out contigs with a low
number of mapped read (less than 10 reads by default).
corset也利用read的信息去过滤低reads支持的contigs,默认少于10的被过滤掉
Corset’s approach is in contrast to other tools used for
clustering contigs as the majority of other tools only use
the sequence information from the assembly.
corset的方法与其他从组装中使用序列信息作为主要的工具去聚成contigs的工具形成对比
Corset works by clustering contigs based on shared
reads, but separates contigs when different expression
patterns between samples are observed
corset基于共享reads进行聚类,但当样品间的表达模式不同的时候进行分离
This is implemented using an agglomerative hierarchical clustering
algorithm.
这是用一种凝聚层次算法实现的
The distance between any two contigs is
defined in relation to the number of reads that are shared
between contigs, such that a lower proportion of shared
reads results in a larger distance (see Materials and
methods).
任何两个重叠群的距离定义成与重叠群中的reads的数量有关
such that a lower proportion of shared
reads results in a larger distance (see Materials and
methods).
比如重叠群更低比例的共享reads导致更大的距离
Genes that share sequence, such as paralogues,
are likely to have small distances, as many reads are
shared.
比如有旁系同源的共享reads可能有更小的距离,因为他们之间有很多共享reads
As we do not want these contigs to be clustered,
Corset performs a test to detect whether the relative
expression levels between the pair of contigs is constant
across conditional groups (or experimental groups).
如果我们不希望这些重叠群被聚类,corset会测试检测相对表达水平经过不同的条件组处理在成对的重叠群是否是一致的
If the
relative expression between the two contigs is not con-
stant, the distance between the two contigs is set to the
maximum.
如果相对表达水平在2个条件中是不一致的,contigs的距离设置成最大
This is incorporated into (纳入)the algorithm as a
likelihood ratio test where the null hypothesis assumes
that the ratio between counts from the two contigs are
equal across conditional groups, whereas the alternative
hypothesis allows this ratio to vary with conditional group
这种方式被纳入算法作为一种可能性比率测试在0假设 假定counts在两个重叠群等于条件组,
然而选择假设允许比例随条件组变化
The count data for this contig ratio test are modeled as
Poisson distributed and a P-value threshold of approxi-
mately <10-5 is applied by default (see Materials and
methods for a detailed description and justification of
thresholds).
对count数据的重叠群比例试验使用泊松分布的模型,默认的p值大约是1e-5
The contig ratio test that separates contigs with shared
sequence but differing expression ratios is one of the novel
features of the Corset clustering algorithm.
重叠群试验中分离重叠群包含共享reads但表达率不同,在corset聚类算法中是一种新的feature特点
Although this
feature can be switched off(关掉) - for example, to ensure differ-
entially spliced isoforms are clustered together - we find it
is effective in separating contigs from different genes
(Additional file 1: Figure S7).还没看
尽管这种feature能被关掉
例如 ,为了确定不同的剪接可变剪接被聚在一起,我们从不同基因中发现它有效地在分离重叠群中
For example, Figure 2 shows
the human ATP5J and GABPA genes, which reside on(位于)
opposite strands but have overlapping (重叠)UTRs.
2个基因位于相反的链上,但是有UTRs重叠
The assem-
bly of human primary lung fibroblast data produced eight
contigs for this region (see Materials and methods).
选至人肺纤维原细胞 产生8个contigs
While
there are contigs for each of the genes separately (contigs
1 to 3, and 8) the use of a non-stranded protocol results in
contigs with the two genes assembled together (contigs 4
to 6).
每个基因分离的一些重叠群1,3,8....
When the contig ratio test is not implemented, all
these contigs are assigned to the same cluster and no
significant differential expression is detected between the
knock-down and wild-type conditions (false discovery rate
(FDR) = 0.053).

当重叠群率测试没有实行,
所有这些contigs被分配到一样的类别而且在敲除和野生型条件没有明显的差异表达被检测到(FDR 0.053)
However, examining the contig count
ratios between pairs of contigs tells a different story
(Figure 2B).
然而,在成对的contigs中测试重叠群count比率 却有着不同的结果
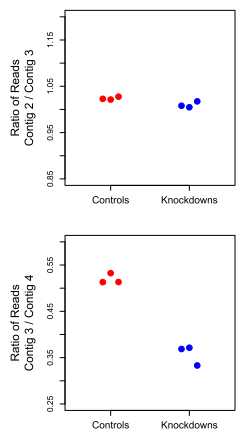
#====================
Figure 2 Corset uses expression information to tease apart contigs from different genes. (A) Assembled contigs from a region of the
human genome containing the two genes ATP5J and GABPA. Trinity assembles 8 contigs (bottom track), which are grouped into one cluster if the
contig ratio test is not applied. Including this test allows corset to separate this region into four clusters (boxes). Notably, contigs 4 to 6 are false
chimeras, caused by the overlapping UTRs of ATP5J and GABPA. These genes are differentially expressed, as shown by base-level coverage, averaged
over replicates (top track). (B) When clustering, Corset checks for equal expression ratios between conditions when calculating distances between pairs
of contigs: here we consider pairs contigs 2 and 3 (top) and contigs 3 and 4 (bottom). The ratio of the number of reads aligning to each contig is
plotted for each sample (dots). It can be seen that contig 2 and contig 3 have the same expression ratio across groups and so are clustered together
while contig 3 and contig 4 have different expression ratios between conditions and so are split. This feature helps Corset separate contigs that share
sequence but are from different genes.
#====================
The count ratios of contig 3 and contig 2
are constant across samples, implying they should be in the
same cluster.
By contrast, the contig ratio between contig 3
and contig 4 is significantly different across conditions and
so Corset splits them into different clusters.
2.3聚在一起,3。4没有聚在一起
When tests for
all pairwise combinations(两两组合) are performed, these eight contigs
are separated into four different clusters and statistical
testing for differential expression reveals cluster a and
d are significantly differentially expressed in opposite
directions (FDR = 10-11 and 10-7, respectively).
当两两组合试验,这8组被分离到4个不同的类并且对差异表达的统计试验揭示 a簇和d簇在相反的方向是显著差异表达的
Once Corset is applied to(应用到) the full dataset the contig
groupings that are representative of genes are reported
and will be referred to henceforth as clusters
一旦corset被应用到所有的数据集,contig组会被当成基因被报道并且后面会被称为簇
Corset also
reports the number of read counts associated with each
cluster.
corset也会报道与每一簇有关的reads counts的数量
All the reads are uniquely assigned to a cluster
(see Materials and methods);
所有的read会唯一比对到cluster
hence, each read is only
counted once, even though the reads were originally
multi-mapped to contigs.
因此,每一个read只被计算一次,及时原始的reads比对到多个contigs
The read counts table can be
supplied (提供给)to count-based differential expression programs
for statistical testing
reads count能提供给基于count的数据做差异表达分析
Testing Corset on model organism datasets
测试corset用于模式物种的数据集
We tested the performance of Corset against other cluster-
ing and counting methods using three RNA-seq datasets:
chicken male and female embryonic tissue [24],
我们用RNAseq 数据集测试corset对于其他的聚类和定量的方式的性能
human
primary lung fibroblasts, with and without a small inter-
fering RNA (siRNA) knock down of HOXA1 [25], and
yeast grown under batch and chemostat conditions [26].
We selected three model organisms in order to compare
our de novo differential gene expression (DGE) results
against a genome-based analysis (referred to herein as the
truth dataset).
人HOXA1 肺细胞敲除和没有敲除siRNA
酵母生长在处理过和恒定的培养基
We selected three model organisms in order to compare
our de novo differential gene expression (DGE) results
against a genome-based analysis (referred to herein as the
truth dataset).
我们选择3种模式物种为了比较我们从头组装不同差异基因的表达结果和基于基因组的组装
(这里引用真实数据集)
In the chicken dataset we tested for DGE
between males and females.
用公鸡母鸡作为DGE测试的数据集
The homology between
chicken genes, which is around 90% on the sex chro-
mosomes [24],
鸡的相似同源基因,有90%在性染色体中
offered a challenging test for clustering
algorithms.
对聚类算法提供了一个有挑战性的测试
The human dataset was selected because
human is one of the best annotated species
是注释最好的物种之一
and the
yeast was used to assess whether clustering is beneficial
for organisms with minimal splicing.
酵母被用来评估聚类对最小可变剪接的物种是否有益
Each dataset was
assembled using Trinity and Oases, which have different
underlying assembly strategies, to ensure that the results
were consistent. Overall, six different assemblies were
used as a starting point for the evaluation of Corset.
为了确保组装结果一致,每个数据集用在组装策略有根本差别的trinity和Oases
总体来说,6中种不同的组装被当做评估corset的出发点
Corset clustering results in a good balance between
precision and recall
corset聚类结果在精确度和完整度都有比较好的平衡
We were initially interested in comparing the clustering
produced by Corset with other available methods.
我们最初对比较corset和其他可以利用的方法产生聚类群的比较感兴趣
Both
Trinity and Oases provide some clustering information
with their output, which is based on the partitioning of
the de Bruijn graphs during the assembly (referred to as
components and locus, respectively).
trinity和Oases在输出中提供一样的聚类群信息,他们在组装策略都是基于deBrujin图(在这里分别被成为成分和轨迹)
Standalone tools
based on sequence similarity are also frequently used
[27,28], with CD-HIT-EST a popular choice [29,30].
单独基于序列相似度的工具【27.28】和流行的cd-hit-est【29.30】也被经常使用
比较了TGICL,BLAT,CD-HIT-EST
TGICL和 和cd-hit的组装策略一样?

We evaluated Corset’s clustering against CD-HIT-EST
and the assemblers’ own clustering. For chicken, over
300,000 contigs were assembled while for human, over
100,000 contigs were assembled (Table 1). A large number
of clusters were reported by Trinity and CD-HIT-EST -
for example, over 200,000 clusters on the chicken dataset.
By default, Corset removes contigs with a very low num-
ber of reads supporting them, to give fewer clusters in all
cases (Additional file 1: Table S1). This makes the cluster
list more manageable, without compromising sensitivity
corsetjiedu

![[c++基础]STL](https://img.php1.cn/3c972/1edc8/c5a/a04ecc977fd8ed28.jpeg)



 京公网安备 11010802041100号 | 京ICP备19059560号-4 | PHP1.CN 第一PHP社区 版权所有
京公网安备 11010802041100号 | 京ICP备19059560号-4 | PHP1.CN 第一PHP社区 版权所有