近日,百度在国际顶级期刊《Nature》旗下子刊《Nature Machine Intelligence》(Impact score 16.65)上发表了AI+生物计算的最新研究成果《Geometry Enhanced Molecular Representation Learning for Property Prediction》,提出“基于空间结构的化合物表征学习方法”,即“几何构象增强AI算法”(Geometry Enhanced Molecular Representation Learning,GEM模型),揭示了一种基于三维空间结构信息的化合物建模方法,以及在药物研发中的应用。
论文链接:
https://www.nature.com/articles/s42256-021-00438-4
公开资料显示,《Machine Intelligence》是《Nature》旗下专注于机器学习领域的顶级期刊,过去两年该期刊的影响因子已超过16。在该项研究中,百度螺旋桨PaddleHelix团队首次将化合物的几何结构信息引入自监督学习和分子表示模型,并在下游十多项的属性预测任务中取得SOTA,成为百度在AI赋能药物研发领域对外公开的又一项重磅成果。
药研领域求变,AI+生物计算成最佳选择
众所周知,药物研发的成本高、周期长、风险高。据美国塔夫茨大学2014年的研究,新药进入市场的平均成本约为26亿美元,从首次合成到进入临床试验的平均耗时为31.2个月,从一期临床到上市长达96.8个月。另一方面,随着全球迈入老龄化社会,对新药的需求也在逐年增加,到2024年全球医药市场总规模将超过11万亿。与之相反,制药公司每10亿美元投资所获得的上市新药数量却在逐年下降。如何通过新的技术手段,快速找到有潜力的候选药物,降低进入临床试验失败的风险,就成为药物研发领域最亟需解决的问题。
在计算方法出现之前,药物研发基本通过生物实验的方法来寻找药物,成本高昂且耗时长,随着计算化学和计算生物学的发展,也有通过传统机器学习方法辅助进行药物设计的,但这些方法或多或少在效果和效率层面有不足,以小分子为例,要找到一个候选药物,筛选(搜索)的数量级达到10的60次方,传统计算方法很难高效完成。另一方面,随着AI技术的发展和普及,药物研发也逐渐进入到AI时代,天生擅长处理大数据的AI深度学习技术,就成为近年来大家关注的焦点,希望通过AI新技术提升药物研发效率,减少后期失败概率,降低药物研发成本。
化合物的性质预测的主要目的在于及时发现理化性质不达标的化合物,以降低候选化合物进入临床实验失败的风险,提升药物研发的成功率。传统的化合物性质预测分析一般采取实验方式,成本高昂且耗时长。业内也有一些基于AI算法的工作,但大多是使用化合物的二维信息,没有纳入化合物的三维空间结构信息。而百度首次提出,将化合物的空间结构信息引入到化合物预训练中,通过几何增强的自监督学习,对化合物分子进行表征,通过化合物的表征自主推断出空间结构信息,进而预测化合物分子的性质属性,以辅助进行药物研发,提升效率,降低成本。
值得一提的是,该研究由百度螺旋桨PaddleHelix生物计算团队独立完成,并已经在药物研发领域,携手合作伙伴在早期药物研发管线中落地。
百度GEM模型加速药物研发进程
很多的研究工作都证明了机器学习技术,特别是深度学习在化合物性质预测方面的巨大潜力,这些工作使用序列(SMILES表达式)或是图(原子为节点,化学键为边)来表示化合物,用序列建模或者图神经网络(GNN)去预测化合物的属性。有些研究直接把每个化合物看作一个图,利用基于图拓扑结构的自监督学习方法进行分子表征,比如,遮盖并还原化合物图中的原子,化学键或子结构。但是,这些方法都只把化合物视为拓扑图,没有充分利用化合物的几何结构信息。而化合物的几何结构,即三维空间结构,对化合物的物理,化学,生物等性质都起着关键性的作用,具有相同拓扑结构的两个化合物的空间结构可能完全不同。另一方面,由于生物实验复杂的操作和高昂的成本,化合物的标注数据十分稀少且珍贵。稀疏的数据让深度神经网络极易过拟合,难以发挥强大的建模能力,如何从海量的无标注化合物中学习高质量的化合物表征成为化合物建模和属性预测的关键。
鉴于此,百度提出一种全新的基于空间结构的化合物建模方法——几何构象增强AI算法GEM,并设计了多个几何级别的自我监督学习策略,用于学习化合物的空间结构知识,使得化合物的表征能自主推断出空间结构信息。这项技术在十多个基准的化合物属性预测数据集上均取得出色成绩,并成功应用到候选化合物的ADMET成药性预测任务上,取得良好收益。
解读几何构象增强AI算法GEM模型
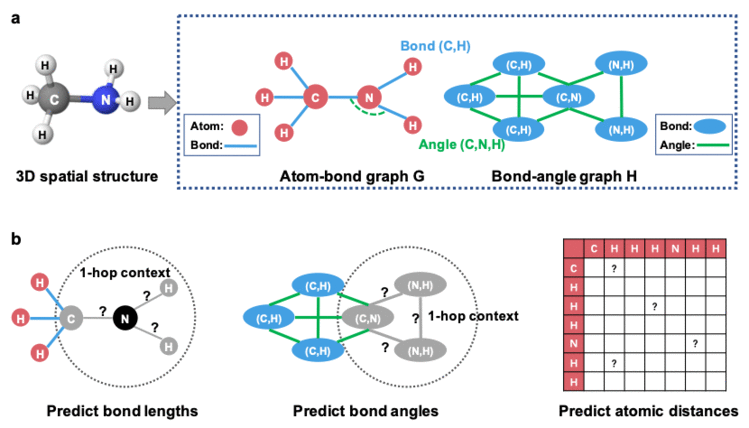
几何构象增强AI算法GEM模型包含两个主要部分:基于空间结构的图神经网络(a)和多个几何级别的自监督学习任务(b)。
图1: GEM的整体框架
1. 基于空间结构的图神经网络
由于化合物的集合结构可以完全被原子-化学键键长-键角确定。GEM提出了一种基于空间结构的图网络,同时对原子-化学键-键角的关系建模空间结构信息。每个化合物由两个图组成:原子-化学键的图G和化学键-键角的图H。类似于过往的工作,原子-化学键的图G以原子作为图的节点,化学键作为连接原子的边。而化学键-键角的图H则为首次引入,以化学键作为图的节点,两个化学键所形成的键角为图的边。图神经网络包含多轮迭代,而化学键作为每一轮迭代中图G和图H的桥梁进行信息互通。最后一轮迭代的表征被用于化合物属性预测。
2. 基于空间结构的自监督学习
为了使模型更好学习到化学空间知识,GEM不单单只是将几何信息作为输入,更进一步地设计了基于几何信息的学习任务(目标):预测化学键的长度;预测化学键组成的键角;预测两两原子之间的距离。其中,键长和键角描述化合物的局部结构,而两两原子之间的距离更关注化合物的全局结构。描述局部结构的自监督学习任务随机挑选化合物中以某个原子为中心的子图并进行遮盖,预测被遮盖的子图中的化学键的键长和化学键间形成的键角。描述全局结构的自监督学习任务则预估原子距离矩阵中的元素。通过这些基于空间结构的自监督学习任务,图神经网络能够有效推断出化合物的空间信息,从而对化合物的表征带来正向影响。
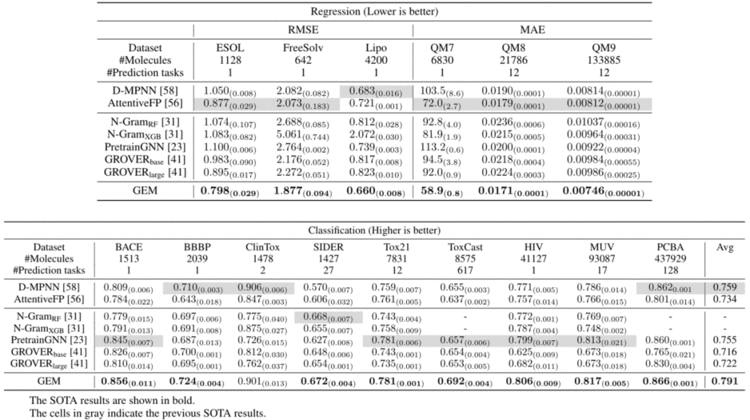
3. 实验结果
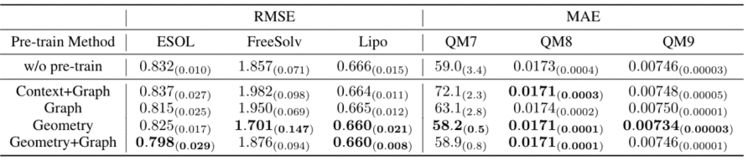
GEM在14个化合物属性的基准数据集中取得了最佳表现,这些数据集都是目前学术界公认的化合物属性预测数据集。例如,在毒性相关的数据集(tox21、toxcast)和HIV(艾滋病)病毒数据集上,GEM预测结果远优于其他baseline模型。总体而言,百度的GEM模型,在ESOL、FreeSolv等回归任务上相对现在方法提升8.8%,在BACE、BBBP、SIDER等分类任务上相对提升4.7%。此外,在自监督学习方法上的消融实验也证明了基于空间结构的自监督学习方法的有效性。
落地ADMET成药性预测和药物筛选等场景
几何构象增强AI算法GEM,能很好的学习化合物的空间结构知识,自主推断出空间结构信息,从而准确地预测候选化合物的ADMET性质——吸收(Absorption)、分布(Distribution)、代谢(Metabolism)、排泄(Excretion)和毒性(Toxicity),帮助在药物研发早期快速筛选潜在成功率更高的化合物。据了解,百度的该项研究已经应用于药物研发领域,在合作伙伴的早期药物筛选管线中实现了商业化落地。
此外,几何构象增强AI算法GEM还在药物虚拟筛选和药物联用方面,也起到关键作用。药物虚拟筛选是药物研发的重要一环,旨在通过从大规模的虚拟化合物库中找到与目标靶点有强亲和力的候选化合物。药物联用是通过预测两个药物在不同细胞系中的协同效用,以帮助找出给定药物在某一细胞系内中具有最佳协同作用。使得两种有协同效用的药物能够在保证治疗效果的同时,减少抗药性的产生。并通过降低使用剂量提升药物的安全性。
关于百度螺旋桨PaddleHelix
螺旋桨PaddleHelix是基于百度飞桨深度学习框架打造,面向新药研发、疫苗设计、精准医疗等场景的生物计算平台,为生物医药领域的研究者提供全面的AI+生物计算的模型工具和技术方案。目前,螺旋桨PaddleHelix平台已开放多个模型,涵盖分子生成、虚拟筛选、ADMET预测、蛋白/RNA结构预测、mRNA序列设计、双药联用等方面。
除此之外,在PPI蛋白-蛋白相互作用,组学的表征和精准用药等方面,螺旋桨PaddleHelix团队也开展了相关的工作,并在多个国际竞赛中取得佳绩,相关研究成果也将陆续开放给大家体验试用。未来,螺旋桨PaddleHelix生物计算平台,还将继续秉持开源开放的态度,继续携手合作伙伴赋能生物计算行业,共建AI+生物计算的生态和服务。
基于空间结构的化合物表征学习方法GEM已通过螺旋桨PaddleHelix平台对外开放,欢迎大家使用。
GitHub地址:
https://github.com/PaddlePaddle/PaddleHelix
平台地址:
https://paddlehelix.baidu.com/
合作洽谈:
baidubio_cooperate@baidu.com
相关推荐
AI+Science:基于飞桨的AlphaFold2,带你入门蛋白质结构预测
RNA结构预测竞赛圆满落幕,优胜选手花式解题思路齐奉上
图学习+分子表示学习,飞桨刷新药物分子性质预测榜单
关注【飞桨PaddlePaddle】公众号
获取更多技术内容~











 京公网安备 11010802041100号
京公网安备 11010802041100号