欢迎关注”生信修炼手册”!
通过GWAS分析可以识别到与性状关联的SNP位点,然而从生物学角度出发,我们更想了解的是哪些基因或者通路导致了这些位点与性状的关联现象。为了解决这一问题,科学家们发明了DEPICT这款软件,通过预测基因的功能来对GWAS的结果进行解释。
通过该软件, 可以进行以下几种分析
通过基因功能对gwas结果中涉及到的基因进行重要性的排序,也就是所谓的gene prioritization
对显著关联位点对应的基因进行功能富集分析
对显著关联位点对应的基因进行组织/细胞表达特异性分析
一句话总结,就是从基因对应的功能,pathway以及表达的位置来解释GWAS结果。相关的文章发表在nature communications上,链接如下
https://www.nature.com/articles/ncomms6890
官网如下
https://data.broadinstitute.org/mpg/depict/
其分析过程示意如下

可以分为以下4大步骤
第一步,预测关联位点对应基因的功能。首先利用77840个样本的芯片数据,构建了一个基因共表达网络,属于一个module的基因认为具有相似的功能。对于显著位点,根据LD程度确定该位点的筛选范围,将对应范围内的基因都认为是候选基因,对于其中功能未知的基因,利用与该基因共表达的已知功能基因来确定其功能
第二步,候选基因功能富集分析,通过构建的14461个基因功能数据集来进行功能富集分析
第三步,候选基因表达特异性分析,同样利用富集分析的算法,来分析候选基因在哪些组织或者细胞中高度表达
第四步,gene prioritization, 利用上述的功能预测和富集分析结果,对候选基因进行重要性排序
该软件所有内置的数据都包含在了源代码包中,所以源码包有2.9G之大,直接下载即可使用。用法如下
1. 自定义脚本参数
该软件的可执行脚本名为depict.py, 修改参数需要直接编辑这个文件,在软件的开头有部分代码需要自定义,示意如下

通常只需要修改前3个参数即可,param_analysislabel指定输出结果文件的前缀,path_snpfile指定gwas结果中显著关联的SNP位点,每行为一个rs号,path_results指定输出结果所在的文件夹。
2. 运行脚本
参数定义好之后,直接运行该脚本即可,代码如下
python depict.py
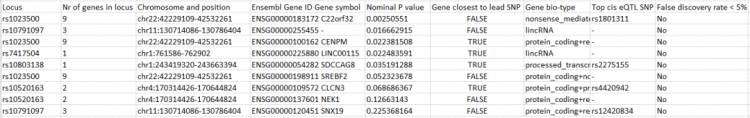
输出结果为多个文件,后缀为loci.txt文件保存了SNP位点对应的基因信息,内容示意如下

后缀为genesetenrichment.txt的文件保存了候选基因的基因集富集分析结果,内容示意如下

后缀为tissueenrichment.txt的文件保存了候选基因的组织/细胞富集分析结果,内容示意如下

后缀为geneprioritization.txt的文件保存了候选基因的排序结果,按照p值从小到大排序,内容示意如下
和MAGMA等gene-based关联分析工具不同,DEPICT提供了一种新的思路,来识别gwas结果中值得深入挖掘的候选基因,并对这些基因的重要性进行排序。
·end·
—如果喜欢,快分享给你的朋友们吧—
往期精彩
自己动手进行逻辑回归,你也可以!
GWAS大家都知道,Gene-Based GWAS你了解吗?
3步搞定GWAS中的Gene Set Analysis
你听说过Epistasis吗?
GWAS中的Gene-Gene Interactions如何分析?看这里
终于搞清楚了Lasso回归和Ridge回归的区别
odd ratio置信区间的计算,你学会了吗?
多元回归分析存在多重共线性了怎么办?
基因型与表型的交互作用如何分析,多元回归来搞定
曼哈顿图就够了吗?你还需要LocusZoom
GWAS做完了,下一步做什么?
GWAS meta分析
GWAS样本量不够怎么办,meta分析了解一下
你没看错,搞定GWAS meta分析只需一行代码!
meta分析的森林图不会画?看这里
GWAMA:GWAS meta-analysis的又一利器
点击鼠标即可完成GWAS meta分析,任何人都可以!
用R进行gwas meta分析,原来如此简单
基因型填充
GWAS中的genotype imputation简介
基因型填充中的phasing究竟是什么
基因型填充前的质控条件简介
使用shapeit进行单倍型分析
gtool:操作genotype data的利器
使用IMPUTE2进行基因型填充
使用Beagle进行基因型填充
使用Minimac进行基因型填充
使用Eagle2进行单倍型分析
X染色体的基因型填充
文献解读|不同基因型填充软件性能的比较
Haplotype Reference Consortium:最大规模的单倍型数据库
Michigan Imputation Server:基因型填充的在线工具
CNV分析
aCGH芯片简介
aCGH芯片分析简介
基于SNP芯片进行CNV分析中的基本知识点
PennCNV:利用SNP芯片检测CNV
DGV:人类基因组结构变异数据库
dbvar:染色体结构变异数据库
DGVa:染色体结构变异数据库
CNVD:疾病相关的CNV数据库
DECIPHER:疾病相关的CNV数据库
全基因组数据CNV分析简介
使用CNVnator进行CNV检测
使用lumpy进行CNV检测
CNVnator原理简介
WES的CNV分析简介
XHMM分析原理简介
使用conifer进行WES的CNV分析
使用EXCAVATOR2检测WES的CNV
靶向测序的CNV分析简介
使用CNVkit进行CNV分析
DECoN:最高分辨率的CNV检测工具
TCGA
TCGA数据库简介
使用GDC在线查看TCGA数据
使用gdc-client批量下载TCGA数据
一文搞懂TCGA中的分析结果如何来
通过GDC Legacy Archive下载TCGA原始数据
使用GDC API查看和下载TCGA的数据
使用GDC下载TCGA肿瘤患者的临床信息
使用TCGAbiolinks下载TCGA的数据
使用TCGAbiolinks进行生存分析
使用TCGAbiolinks分析TCGA中的表达谱数据
使用TCGAbiolinks进行甲基化和转录组数据的联合分析
Broad GDAC:TCGA数据分析中心
使用cBioPortal查看TCGA肿瘤数据
UCSC Xena:癌症基因组学数据分析平台
GEPIA:TCGA和GTEx表达谱数据分析平台
TANRIC:肿瘤相关lncRNA数据库
SurvNet:基于网络的肿瘤biomarker基因查找算法
TCPA:肿瘤RPPA蛋白芯片数据中心
TCGA Copy Number Portal:肿瘤拷贝数变异数据中心
生存分析
生存分析详细解读
用R语言进行KM生存分析
使用OncoLnc进行TCGA生存分析
用R语言进行Cox回归生存分析
使用kmplot在线进行生存分析
肿瘤数据库
ICGC:国际肿瘤基因组协会简介
HPA:人类蛋白图谱数据库
Oncomine:肿瘤芯片数据库
ONGene:基于文献检索的肿瘤基因数据库
oncomirdb:肿瘤相关的miRNA数据库
TSGene:肿瘤抑癌基因数据库
NCG:肿瘤驱动基因数据库
mutagene:肿瘤突变频谱数据库
CCLE:肿瘤细胞系百科全书
mSignatureDB:肿瘤突变特征数据库
GTEx:基因型和基因表达量关联数据库
肿瘤免疫和新抗原
Cancer-Immunity Cycle:肿瘤免疫循环简介
TMB:肿瘤突变负荷简介
肿瘤微环境:Tumor microenvironment (TME)简介
肿瘤浸润免疫细胞量化分析简介
使用EPIC预测肿瘤微环境中免疫细胞构成
TIMER:肿瘤浸润免疫细胞分析的综合网站
quanTIseq:肿瘤浸润免疫细胞定量分析
The Cancer Immunome Atlas:肿瘤免疫图谱数据库
肿瘤新抗原简介
TSNAdb:肿瘤新抗原数据库
使用NetMHCpan进行肿瘤新抗原预测分析
Hi-C数据分析
chromosome-territories:染色质疆域简介
chromosome conformation capture:染色质构象捕获技术
3C的衍生技术简介
解密Hi-C数据分析中的分辨率
A/B compartment:染色质区室简介
TAD:拓扑关联结构域简介
chromatin loops:染色质环简介
Promoter Capture Hi-C:研究启动子区染色质互作的利器
使用HiCUP进行Hi-C数据预处理
Juicer:Hi-C数据处理分析的利器
Juicer软件的安装详解
Juicebox:Hi-C数据可视化利器
Juicer实战详解
HiC-Pro:灵活的Hi-C数据处理软件
HiC-Pro实战详解
3D Genome Browser:Hi-C数据可视化工具
HiCPlotter:Hi-C数据可视化工具
3CDB:基于3C技术的染色质互作信息数据库
3DIV:染色质空间互作数据库
4DGenome:染色质相互作用数据库
4D nucleome project:染色质三维结构研究必不可少的参考项目
3dsnp:SNP在染色质环介导的调控网络中的分布数据库
iRegNet3D:疾病相关SNP位点在三维调控网络中的作用
使用WashU Epigenome Browser可视化hi-c数据
HiGlass:高度定制的Hi-C数据可视化应用
Hi-C Data Browser:Hi-C数据浏览器
使用FitHiC评估染色质交互作用的显著性
使用TADbit识别拓扑关联结构域
使用pyGenomeTracks可视化hi-c数据
hi-c辅助基因组组装简介
文献解读|使用hi-C数据辅助埃及伊蚊基因组的组装
chip_seq数据分析
Chip-seq简介
chip_seq质量评估之计算样本间的相关性
chip_seq质量评估之查看抗体富集效果
chip_seq质量评估之PCA分析
chip_seq质量评估之coverage分析
chip_seq质量评估之FRiP Score
chip_seq质量评估之cross correlation
chip_seq质量评估之文库复杂度
depth, bedgraph, bigwig之间的联系与区别
bigwig归一化方式详解
使用igvtools可视化测序深度分布
使用UCSC基因组浏览器可视化测序深度分布数据
使用deeptools查看reads分布特征
使用phantompeakqualtools进行cross correlation分析
blacklist regions:NGS测序数据中的黑名单
MACS:使用最广泛的peak calling软件之一
MACS2 peak calling实战
使用SICER进行peak calling
使用HOMER进行peak calling
peak注释信息揭秘
PAVIS:对peak区域进行基因注释的在线工具
使用UPORA对peak进行注释
使用GREAT对peak进行功能注释
annoPeakR:一个peak注释的在线工具
使用ChIPpeakAnno进行peak注释
使用ChIPseeker进行peak注释
使用PeakAnalyzer进行peak注释
使用homer进行peak注释
利用bedtools预测chip_seq数据的靶基因
motif
关于motif你需要知道的事
详解motif的PFM矩阵
详解motif的PWM矩阵
使用WebLogo可视化motif
使用seqLogo可视化motif
使用ggseqlogo可视化motif
MEME:motif分析的综合性工具
使用MEME挖掘序列中的de novo motif
使用DREME挖掘序列中的de novo motif
使用MEME-ChIP挖掘序列中的de novo motif
chip_seq数据库
ENCODE project项目简介
FactorBook:人和小鼠转录因子chip_seq数据库
ReMap:人类Chip-seq数据大全
IHEC:国际人类表观基因组学联盟
Epifactors:表观因子数据库
GTRD:最全面的人和小鼠转录因子chip_seq数据库
ChIP-Atlas:基于公共chip_seq数据进行分析挖掘
Cistrome DB:人和小鼠的chip_seq数据库
chipBase:转录因子调控网络数据
unibind:human转录因子结合位点数据库
chip_seq在增强子研究中的应用
DENdb:human增强子数据库
VISTA:人和小鼠的增强子数据库
EnhancerAtlas:人和小鼠的增强子数据库
FANTOM5:人类增强子数据库
TiED:人类组织特异性增强子数据库
HEDD:增强子疾病相关数据库
HACER:human增强子数据库
SEdb:超级增强子数据库简介
dbSUPER:人和小鼠中的超级增强子数据库
dbCoRC:核心转录因子数据库
使用ROSE鉴定超级增强子
18年文章目录
扫描下方二维码,关注我们,解锁更多精彩内容!

生物信息入门
只差这一个
公众号




 京公网安备 11010802041100号
京公网安备 11010802041100号